In this project, we present a hardware accelerator for QBiC-Pred, an open-source software package to predict transcription factor binding of short DNA sequences. We analyzed QBiC-Pred and found a candidate kernel for hardware acceleration. We implemented a hardware accelerator with 128 lookup tables and 2 processing lanes with a peak computational throughput of 11.4 MM/s (Mega-Mutations per second). Our hardware accelerator delivers a kernel speedup up to 738× compared to our optimized software implementation and reduces the cost per billion mutations by 99.5%.
Characterization and Optimization of the Software Baseline
The initial version of the software was profiled and refactored to identify kernels suitable for FPGA acceleration. The original code was tested on an AWS F1 instance and after intermediate file i/o optimizations and vectorization, a speedup of 152× was observed over the original software using a limited list of transcription factors and 1 million simulated mutations. More software optimizations are available but are not further explored in the context of this project. We will refer to the improved software version as V3, which will serve as our baseline for the remainder of this project.
We profiled V3 of the software in which three main phases are identified: initialization, isBound (our kernel of interest), and result writing. The isBound phase, our kernel of interest, can be divided into three parts: isBound (the actual computations), post-processing, and other overhead (e.g. mutation list filtering). With V3 of the software, we were able to profile to 32 transcription factors and 1 million mutations using an AWS c5n.4xlarge instance. The c5n.4xlarge instance is de- signed for compute-intensive tasks and provides a reasonable number of vCPUs (16) and high-performance storage. Our application benefits most from fast disk i/o and more vCPUs while memory size is not a primary performance constraint. Hence, the c5n.4xlarge instance matches the demands of our applications well. The total runtime for different configurations can be found in Figure 1 and the breakdowns of runtime for 1 million mutations can be found in Figure 2.
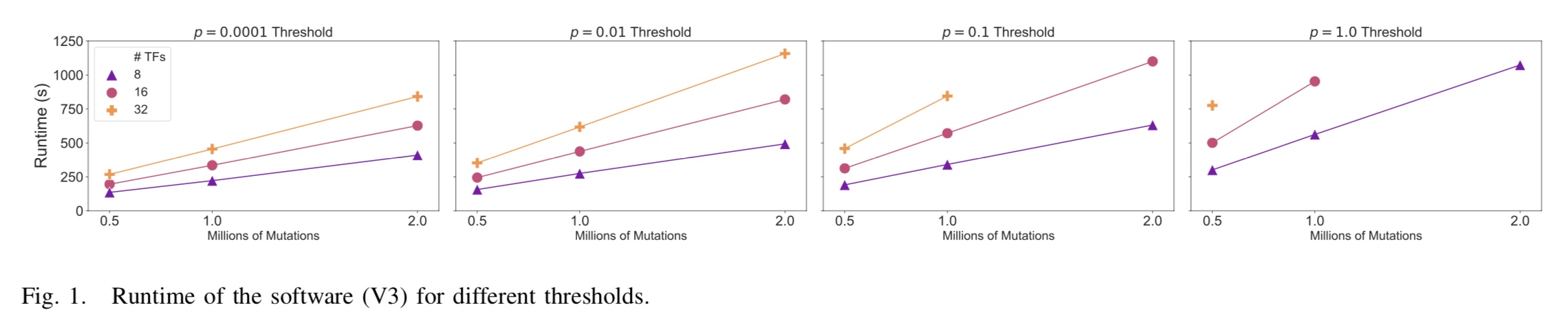
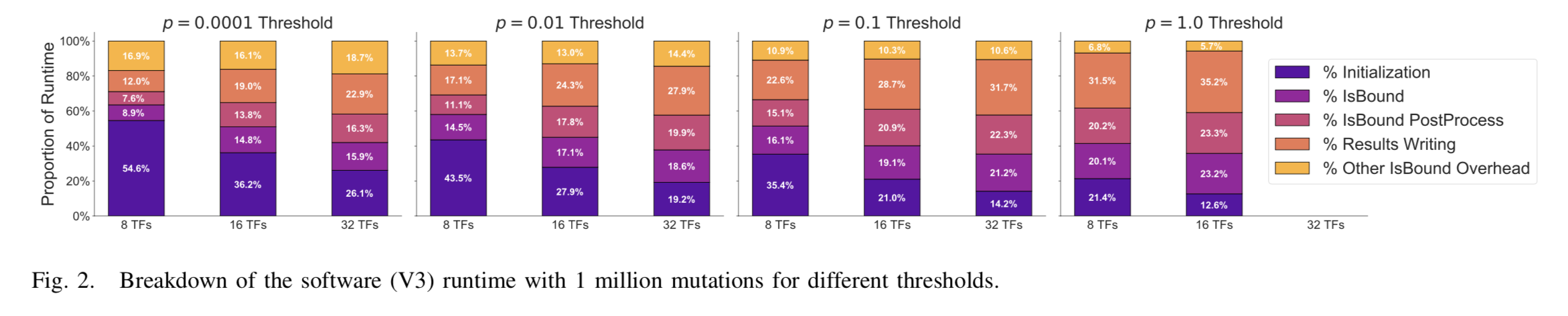
Figure 1 clearly shows that the application runtime increases when the number of transcription factors or the number of mutations increases. The runtime increases proportionally to the number of mutations to process while increasing the number of transcriptions factors results in a sub-linear increase of the runtime. The number of mutations scales all application phases proportionally while the number of transcription factors only scales some application phases proportionally. A lower p-value threshold results in more mutations being filtered. Hence, the runtime increases when the p-value threshold increases.
The breakdown of V3 runtime for 1 million mutations, shown in Figure 2, clearly shows that when the computational demand increases (i.e. more transcription factors and more mutations to process) the kernel and result writing phases dominate runtime. Result writing takes a lot of time because results are written in verbose CSV format to disk and file i/o performance imposes a bottleneck. Using efficient file formats can minimize the time required for result writing, but this phase is not suitable for hardware acceleration and therefore outside the scope of this project.
Proposed Solution
After analysis of V3, the is_bound_18mer() function (isBound phase) stands out for FPGA acceleration. The execution time of this function scales with the number of transcription factors and the number of mutations to process. Developers of the software informed us the target number of transcription factors would be on order of several hundred (up to 600, probably less), while the number of mutations should be as large as possible.
We analyzed the data dependencies of our kernel function and were able to transform our data flow graph to one more suitable for hardware acceleration. We chose to first compare all elements of the lookup tables to the two thresholds used for the reduction operator, reducing memory requirements for the lookup tables significantly. Since these thresholds do not change very often, lookup tables can be converted once and stored on disk for later reuse. The reduced size lookup tables can be stored in fast on-chip random access memory on the FPGA for data-locality while the large mutation data can be read sequentially from DRAM. By loading many lookup tables to the on-chip memory we can perform calculations on multiple transcription factors for a single mutation in parallel, exploiting earlier identified data-parallelism. When we are processing multiple transcription factors in parallel, we are not able to filter the mutation list based on p-values beforehand. Every combination of mutation and transcription factor will be processed and filtering based on p-value can be performed on the set of results.
System Design
Based on our transformed kernel data flow, we can identify sources of parallelism that can be exploited by a hardware accelerator. Our kernel independently applies the is_bound_18mer() function to every input mutation for every lookup table (transcription factor), which introduces two potential degrees of data-parallelism. Lookup tables are pseudo-randomly accessed and require much less memory than large sets of mutations, therefore we chose to design a streaming architecture that exploits data-parallelism and data-locality by keeping as much as possible lookup tables simultaneous in on-chip BRAM while mutation-data is stored in DRAM and streamed through the data-path of the accelera- tor. Our streaming architecture consists of multiple sequential modules working in parallel leveraging task-level-parallelism. Results are directly written back to DRAM by our architecture. Hence, all our DRAM accesses are now sequential which will benefit practical memory throughout significantly while all random lookup table accesses are handled by BRAM.
The peak computational throughput of our accelerator is limited by the throughput of the Lookup modules. Every mutation requires 18 table lookups which all take a single cycle. The registered BRAM memory output has a latency of 2 cycles and handshaking on the input and output requires at least one cycle each. The total latency of a lookup module, assuming data is presented and the following IsBound module is ready, is therefore 22 cycles. Every lookup module has two lanes and therefore the peak computational throughput of our accelerator is 11.4 MM/s (Mega-Mutations per second). If we assume all lookup tables are active, the resulting peak output bandwidth of our accelerator is 727.3 MB/s. The input bandwidth is not dependent on the number of active tables. With six mutations stored per block, the peak input bandwidth is 60.1 MB/s. The parallel lookup modules can generate a lot of output per input mutation and the input data bandwidth can be amplified up to 12 times when using all 128 available tables.
The number of mutations that can be processed in a single batch (e.g. a single start command) is limited by the DRAM memory size and depends on the number of active lookup tables. When using all 128 lookup tables, we can store up to 230.8 million mutations and results in 16 GB of memory. The lowest amplification factor is found when using 32 or fewer lookup tables simultaneously due to the 32-byte block size for writing to memory. When 32 or fewer lookup tables are active, we can store up to 750 million mutations and results in 16 GB of memory.
Evaluation and Conclusion
Runtimes of the kernel including data transfers to and from the FPGA. Execution times scale linearly when increasing the number of mutations, which is as expected. Kernel runtime should in theory not increase when the number of transcription factors increases (up to 128) because all transcription factors are processed in parallel.
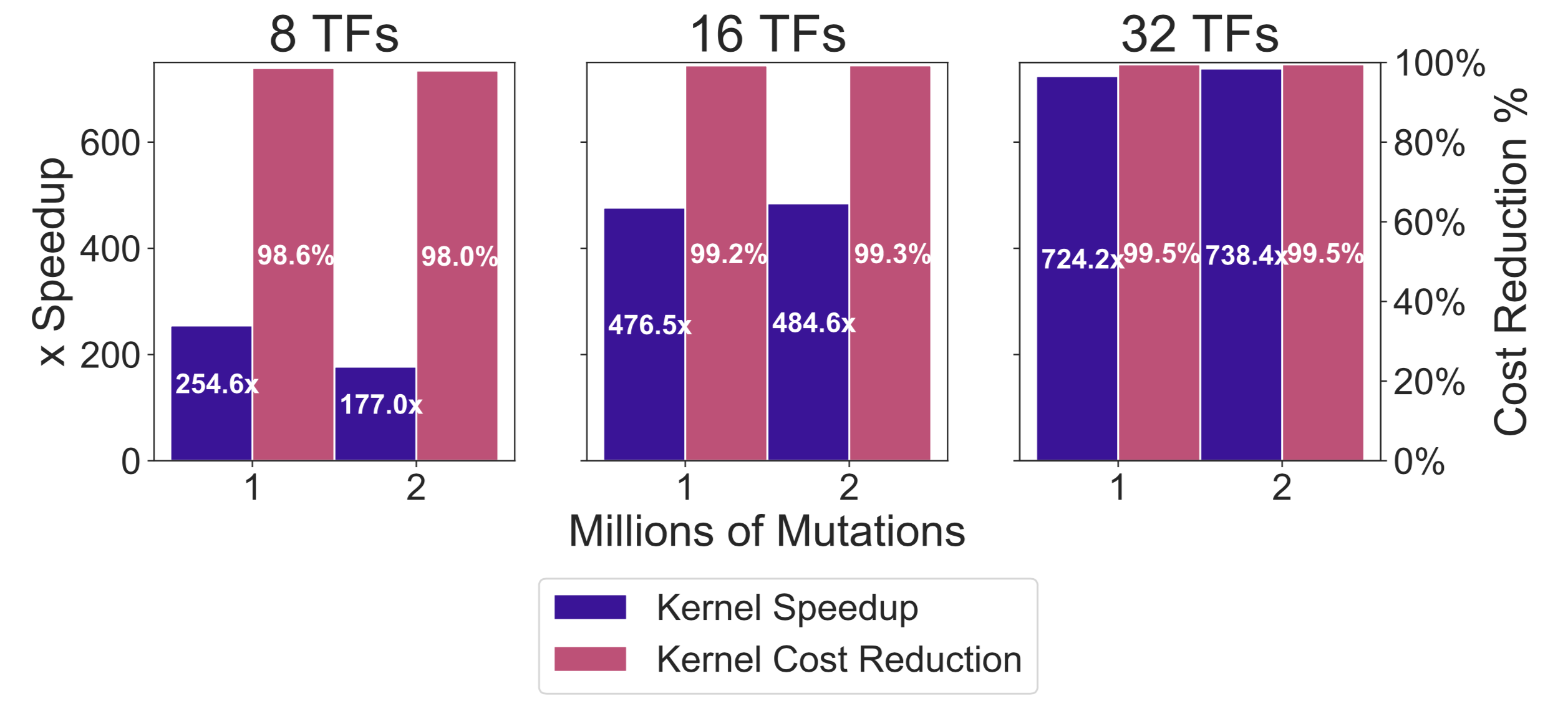
Finally, we calculated the kernel speedup and cost reduction our accelerator delivers compared to the V3 software. Figure below shows that the accelerator mainly benefits when more lookup tables are active, which is the accelerator’s main source of data-parallelism. We observe speedups up to 738× and cost reductions up to 99.5%.